🎄12 Days of HPC 2021
Subclone deconvolution is coming to town!
During the month of December we’re featuring blog posts from researchers from across the University of Leeds showcasing the fantastic work they do using our High Performance Computing system. Follow us @RC_at_Leeds to keep up to date with our 12 days of HPC blog series.
What’s your name?
Lucy Stead
What department do you work in?
Leeds Institute of Medical Research
What research question are you trying to answer?
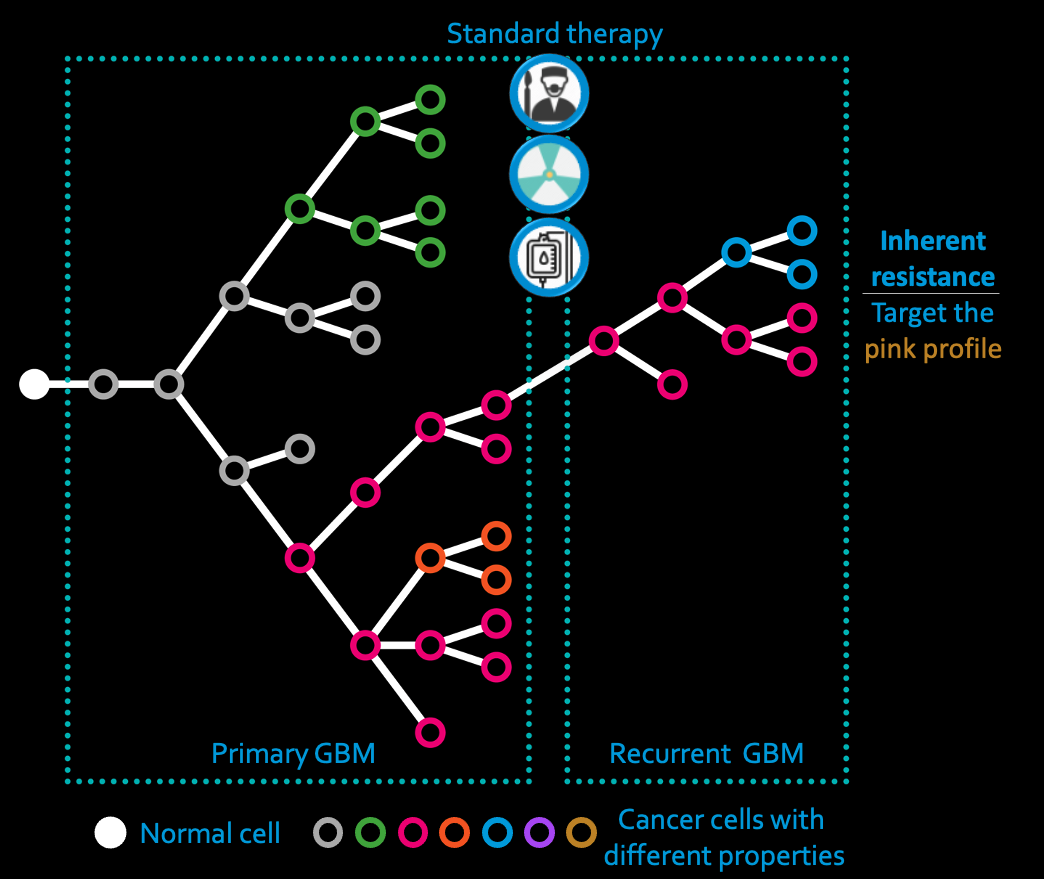
Tumours are made up of subpopulations (or subclones) of cells, each with different mutations that make the cells in that subclone behave differently. This can mean that some of the subclones are able to resist treatment and meaning their continued survival causes cancer recurrence. Think of it like a big ‘ball’ made lego but with the bricks being different colours. If someone came to demolish the ball but the red bricks were really tightly attached together, they might only get so far with destroying it. What we need to do, then, is identify those particular subclones (the red bricks in our analogy) and understand why they, specifically, are treatment resistant and how we can, thus, kill them. This is called subclonal deconvolution and it is HARD. Lots of approaches exit but no-one has ever been able to test them to see a) whether they work, and b) which works best. So this is what Dr Georgette Tanner set our to do. She developed a method that allowed her to grow tumours in silico and then, because she knew every subclone that was present in the artificial tumour, she methodically tested all the methods available for subclonal deconvolution until she could say which methods gave her the right answer!
What tools or technologies do you use in your research? (Programming languages, packages, APIs)
The following open source software was used to create the datasets used in the study: HeteroGenesis (v1.5), w-Wessim2 (v1.0), pblat v652d3b3, ReSeq (v1.2), cutadapt(v1.18), bwa-mem (v0.7.17-r1198-dirty), Picard CollectHsMetrics (v2.19.1-SNAPSHOT-all). The following open source software was used to analyse the datasets: Mutect2, (GATK v4.1.7.0), FilterMutectCalls (GATK v4.1.7.0), Strelka2 v2.9.10, Lancet v1.1, samtools v1.9, bam-readcount v0.8.0, GATK-indel realignment v3.8-1-0, VarScan2 v2.4.4 , scikit-learn v0.23.1, Sequenza v3.0.0, sequenza-utils v3.0.0, TITAN v1.23.1, ichorCNA v0.3.2, HMMcopy v1.26.0, FACETS v0.5.14, cnv_facets v0.15.0, Sclust v1.1, PyClone v0.13.1, PyClone-VI v0.1.0, Ccube v1.0, CNVkit v0.9.6.
How does HPC help your research?
Human DNA is 3 billion letters long, and we need to sequence it to about 100 fold coverage and then we need to analyse all those data simultaneously as the methods learn from all regions of the genome and then we have to apply sophisticated mathematical models and then…. you get the picture: Big data and a shed load of analysis means we need big memory and nodes for days!
What is the potential impact of your research?
Mr Yousef Alghamdi is now using what we have learned to do subclonal deconvolution of paired primary and post-treatment recurrent brain tumours (from the same patient) so we can learn which cell subpopulations survived and why - in the hopes of developing better drugs to treat the tumours. So the impact could be huge: in a nutshell we hope to cure brain cancer!
In your personal opinion what’s the coolest thing about your research?
We sequence the DNA of actual brain tumours from actual patients who are suffering the most horrendous disease. Then we turn it into sequence data that teaches us about this b*stard cancer cells and why they have gone rogue. What we do could one day stop a patient hearing ‘there is no hope’.
In your opinion, what is the ultimate Christmas song?
Walking in a Winter Wonderland
Postscript
Check out more about us here: https://braincancer.leeds.ac.uk/glioma-genomics/ and follow us on Twitter: @GliomaGenomics